
Dosing your degrader. To be or not to be.....oral. That is the question.
27th February 2024
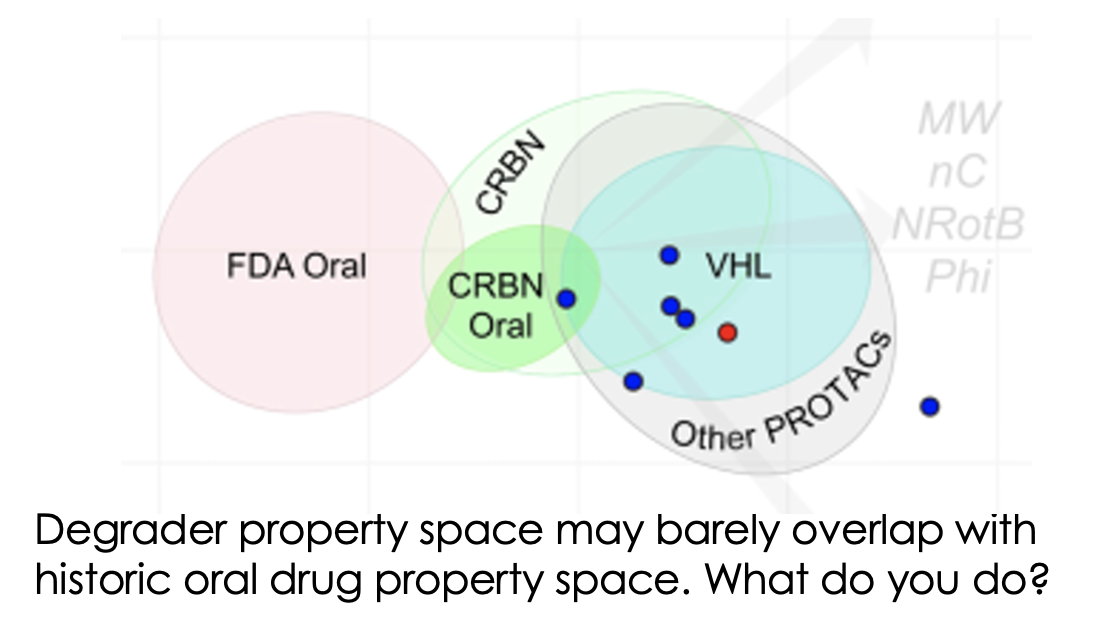
Much has been written about how the properties of bifunctional agents including PROTACs are far outside the “usual small molecule space”. However, now we’re in the current renaissance of small molecule therapeutics including bifunctionals, peptides, macrocycles, conjugates etc, the infamous Lipinski Rule of 5 is feeling more like a mnemonic from a bygone age. Prompted by the recent Caron/Kihlberg Drug Discovery Today article on predicting how to design orally active PROTACs, just how important are the properties of degraders in determining what dosing routes are available to you? And indeed is oral dosing always preferable?
Conventional small molecule drug discovery wisdom will tell you that patients prefer oral tablets for their ease of dosing though, of the 20 best selling drugs of 2022, only 40% of them are orally dosed such has been the recent emergence of biologic therapies (and some other COVID-19 antivirals). Low patient compliance with oral therapies also remains a huge issue with some estimating that up to 50% of all doses of chronic oral treatments may be missed by patients for reasons including forgetfulness, worry about potential side effects as well as low motivation due to perceived lack of immediate efficacy (most drug treatments don’t give you immediate feedback on their efficacy like when you take an aspirin and your headache goes away).
Once a day doses are best, though still not great, but once you ask patients to take your drug twice, three times or more per day, adherence really drops off. This is particularly disappointing for drug discoverers who work tirelessly over many years to get drugs to patients only to find out that many patients who can benefit greatly, simply do not take the treatment, even when they have the chance to. There are many other deeply psychological reasons for drug non-compliance (and suggested potential ways & technologies to improve it) but that’s a topic for another day.
With dosing intervals of most antibody-based therapies typically in the 2-3 week range, this can be a very attractive dosing regimen. Admittedly, if you need to travel to hospital for an intravenous infusion, this can be less appealing to many but with increasing numbers of self-administered subcutaneous or intramuscular devices which can be used at home now available, parenteral dosing routes are becoming more accessible all the time. With needle phobia affecting up to 20% of patients, advances in autoinjector pen technology, where the needle is barely seen, will increase the utility of this route further.
Degrader drugs, especially of long half-life targets, have the potential for extended pharmacodynamic duration of action driven by often slow clearance of drug from target tissue and limited only by the resynthesis rate of the protein. This can give sustained knockdown for often 24h+ after a single dose (see eg here) though whether this will give enough effect to allow once weekly or less frequent dosing will depend on a range of factors and may not be possible for many approaches. Of course, you may not need or want sustained target knockdown to get the right efficacy/safety balance for many degraders so a q7d or q14d etc regimen may still be suitable if you get enough drug to the site of action for long enough even if the target degradation doesn’t persist throughout. Let’s not forget, efficacy is not all about the amount of drug you get on board, it’s whether there’s enough drug in the right place to give the pharmacodynamic effect you need.
Formulation can also have a huge impact – some nice work from the team at GSK showed that a subcutaneous dose of a RIPK2 PROTAC embedded in a Poly-lactic-co-glycolicacid (PLGA) matrix gave controlled drug release maintaining >90% degradation for over 2 months following a single dose in rat. Developing some of these formulations may require a little more work than a standard oral tablet or liquid/powder-filled capsule but may also bring very attractive dosing routes.
Local, non-systemic dosing may also be attractive in some cases and may reduce the risk of tolerability concerns resulting from systemic target degradation. Early clinical data has already been generated with the topical AR PROTAC GT20029 in androgenetic alopecia for example.
If your degrader doesn’t have the pharmacokinetics to allow infrequent dosing paradigms alone then you can always consider the Trojan horse approach of conjugating it to an antibody where the carefully designed conjugate can display the 10-20+ day half-lives often associated with biologics. Indeed the field of degrader-antibody conjugates is growing rapidly (see eg the seminal review and the recent $100M+ deal between Orum & BMS).
Finally of course, if you’re concerned that your bifunctional degrader won’t have a dosing schedule you like in the clinic, molecular glues promise “typical” small molecule pharmacokinetic profiles with good, once daily oral potential. Many groups are following this route which I’ve commented on before so won’t say any more here.
So, let’s return to the topic of how to make your PROTAC (or indeed any bifunctional agent) orally bioavailable when Lipinski-compliant properties are never going to be an option.
The recent DDT article relies on a small dataset only (despite there being huge amounts of oral PROTAC data, almost all of it is not yet in the public domain) but previous analyses from AstraZeneca (eg 2020 review here) or Arvinas (eg 2023 analysis here) also cast light on how to make oral degraders when molecular size approaches the 1kD range.
Is size really important? Well, probably a little as a good, smaller molecule is usually better than a good, bigger molecule but don’t focus on molecular weight as your main med chem goal as you’ll miss most of the relevant optimisation options.
Prevailing wisdom from across the published studies is that predicting oral exposure can be tricky as it is often the result of the contributions of a range of accessible conformations within a shape-shifting 3D ensemble (often termed molecular chameleonicity) which can be hard to pin down or may require complex molecular dynamics or biophysical studies which, for many groups, will be slower and more expensive than just running a few in vivo studies to see what you have. Don’t rely on falling back on your tried and tested small molecule cascade of in vitro DMPK assays (microsomal & hepatic clearance, PAMPA, Caco, MDCK etc) either. These often have no correlation to in vivo bioavailability and half-life - though, if you’re not careful, it may take you a lot of investment in generating in vitro assay data from your own programmes before you learn this. Instead, it’s often best to design molecules with the right kinds of physicochemical properties (ie moderate rigidity in terms of rotatable bonds or use of other conformational biases as well as keeping unsatisfied hydrogen-bond donor count down while keeping the FaSSIF/FeSSIF solubility in a reasonable range) and then just dosing a few in vivo. For poorly orally bioavailable agents, permeability is more often the issue than first pass metabolism so focus on properties which fix that first. Many formulation tricks including use of dispersions, other micronized forms and excipients may also help you increase low bioavailability into a useful range for development.
If you want to have a better chance of seeing oral exposure use mouse (and try cassette dosing to increase the amount of data per study greatly) but rat is really the better, if more challenging, system which may give you more insight into what you may expect in higher species.
Now, in drug discovery we should aim to reduce the number of animals used all the time of course so a strategy of increased in vivo screening my seem counter to this but there are plenty of protocols which make use of small group sizes (which take advantage of the low inter-animal variability usually seen with bifunctional degraders) and truncated profiles which focus on qualitative, directional data rather than a full pharmacokinetic characterisation when in screening mode.
So, to be or not to be oral? There are plenty of non-oral routes to use which are becoming increasingly accessible so it’s worth looking at these, especially if your degrader isn't cereblon-based and so has lower chances of ever being orally available. It may require some extra development work to get the right formulation or dosing regimen but it could be worth it if patients will benefit. If you decide you really must be orally active to compete with existing treatment options then be thoughtful about it, get your potent degraders in the right area of chemical property space (if you can – if not, oral delivery may not be for you) and generate the right in vivo data to guide your optimisation. If you can do this, you’ll stand a good chance of succeeding.

© 2023-2024
All rights reserved. Janus Drug Discovery Consulting Ltd
We need your consent to load the translations
We use a third-party service to translate the website content that may collect data about your activity. Please review the details in the privacy policy and accept the service to view the translations.