
Agonising over the best applications of induced proximity-based therapies
19th January 2024
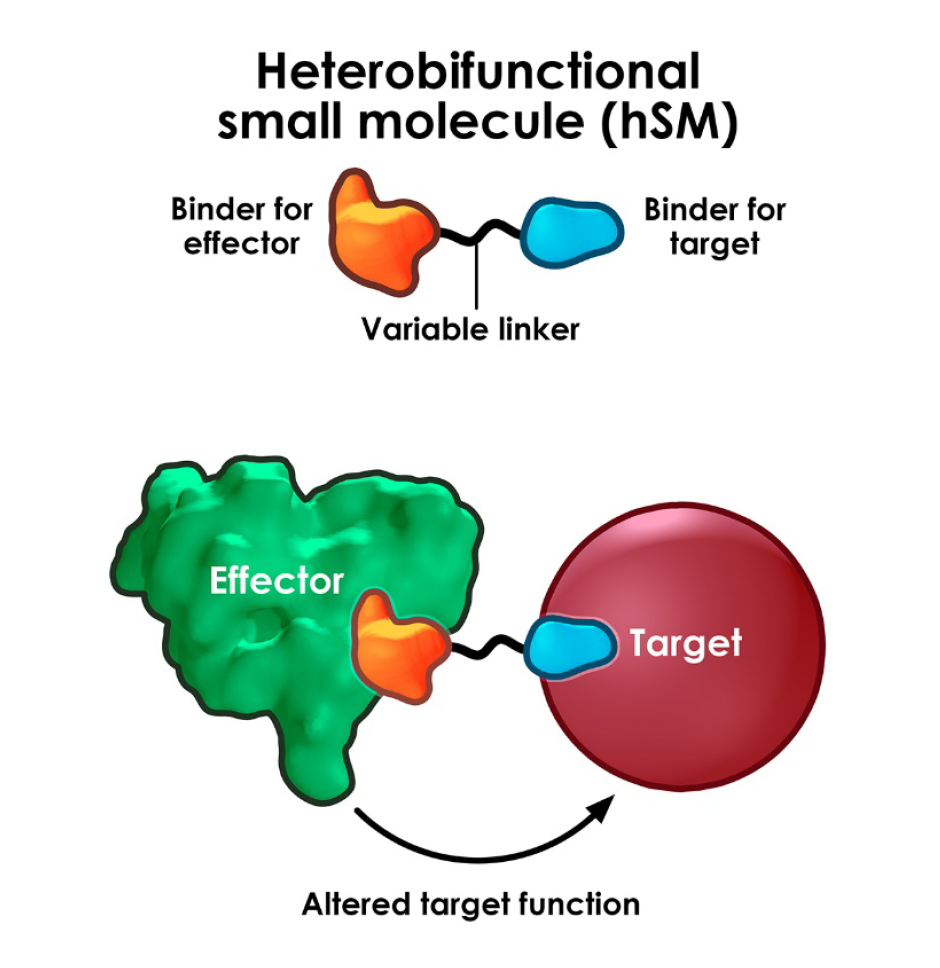
Drug discovery is generally good at identifying therapeutics which stop things working: inhibiting an enzyme, antagonising a receptor, blocking a signalling pathway – great for addressing “gain of function” pathology but sometimes you need to fix or upregulate something that has stopped working in disease (ie “loss of function” pathology). In other words, there’s great scope for therapeutics to agonise or switch on a process.
This is where the advent of induced-proximity-based therapeutics has opened up so much opportunity. By bringing two biological entities, often proteins, into close proximity, new pharmacology can be switched on. This can be via upregulation of existing physiological processes or introduction of completely new ones. Agonism can be powerful and in some cases takes advantage of event-driven or catalytic pharmacology where a transient, induced interaction is sufficient to kick off a biological process from low drug levels.
Although PROTACs & molecular glues are the currently best researched examples of proximity-based therapeutics, where perhaps counter-intuitively, facilitating (agonising) a substrate-E3 ligase interaction leads to functional antagonism and loss of protein function via proteasomal degradation, there are many other areas that induced-proximity is now showing potential. The Amgen team, led by Ryan Potts recently published a very helpful overview of the area which is instructive in the key challenges and hinting where the field could go next.
As well as inducing proximity of pathogenic proteins to ubiquitin E3 ligases (PROTACs & glues), chimeric “x-TAC” agents have now been shown to target disease-relevant proteins to many other “effector” proteins or pathways in a growing list:
- Targeting to the lysosome and autophagosomes for alternative degradation pathways (LYTACs, AUTACs etc - see my previous post on eTPD)
- Targeting to kinases or phosphatases to modify the phosphorylation state of proteins (PhosTACs etc)
- Targeting to induce glycosylation (OGTACs)
- Inducing RNA degradation (RIBOTACs etc – beyond siRNA, CRISPR)
- Stabilising proteins by recruiting deubiquitinases (DUBTACs)
- Acetylation targeting chimera (AceTAGs)
- Recruiting steric blockers (RIPTACs)
- Moving proteins around subcellular compartments (RELOTACs)
Should we expect to see all the new x-TACs pushing into Phase 3 studies as quickly as the prototype PROTACs? Well, maybe for some, but the chimeric molecule game isn’t just plug and play speed-dating to introduce two proteins, there’s reasons why PROTACs work so well and other approaches may require a more thoughtful courtship.
A lot of credit should go to Craig Crews, Ray Deshaies & co-workers for identifying the ubiquitin system as a good prototype for induced-proximity-based pharmacology back in 2001. It has a few things going for it that other systems may not.
Firstly, some ubiquitylating enzymes are not all that choosy about their substrates (A Nobel prize winner once said to me that “If you get an E3 ligase close enough for long enough to its substrate, it will ubiquitylate it!”) so some E3s don’t mind being introduced to new partners. That’s not the case with all of them of course – it’s no accident that, of the 600+ potential E3s, only a handful are actually useful for TPD (E3 ligase selection is a whole other story completely…).
Secondly, it’s been possible to find binders to E3s which don’t completely inhibit the E3 function – this is clearly important as inducing proximity to an inhibited enzyme is unlikely to give you the activity you want. For many proteins however, historic binders are also inhibitors so of less use for use in proximity-based approaches.
Thirdly and obviously, the initial introduction of substrate to E3 ligase may result in ubiquitylation at multiple sites which can then initiate a cascade which ultimately is a one way ticket to destroying the substrate in the proteasome via addition of K48 poly-Ub chains. This is important as the PROTAC may also induce other ubiquitylation events, say addition of mono-Ub or poly-Ub using K63 etc linkages (and we rarely, if ever, characterise this in any detail) which due to the richness of Ub biology, may lead to, not degradation, but a range of other functional consequences including pathway activation which could cause undesired pharmacology & selectivity issues. However, if the substrate is degraded quickly, we may not have to worry about any of these potential “off-degradation” consequences resulting from lack of selectivity in the initial ubiquitylation due to the “self-destruct” nature of any protein which also includes the desired K48 chains.
That’s why E3 ligases & PROTACs may work so well: Many E3s don’t mind taking on new partners and, if the ubiquitylation is not very selective, we may not have to worry as the protein is gone before it has a chance to cause any undesired effects from any unselective ubiquitylation.
Not all induced proximity paradigms may share these advantages however.
Not all proteins may be as permissive to working with new substrates as some E3 ligases. PROTACs likely allow an ensemble of ternary complexes (a subset of which may be active) which in turn may allow ubiquitylation of more than one lysine. We’re getting better at characterising some ternary complexes via x-ray & cryo but in the live cell this is likely more dynamic and complex than we’d want it to be.
However, more broadly, getting two proteins to come together in exactly the right orientation to specifically derivatize a single site is a tough challenge and may require significant effort and design if it is possible at all for some cases. (Glues do this very well for some substrates including some E3s and 14-3-3 proteins for example though rational glue identification for any target has not yet been routinely solved).
So, even if you can get the substrate and effector to interact, getting the specific orientation to selectively give the effect you want may be a challenge. For some effects like deubiquitination, this may be ok but for others, where specificity will be key (eg phosphorylation & dephosphorylation, glycosylation or transcriptional activation), very careful design may be needed. Modification of the substrate at undesired sites may lead to unintended effects which will need to be managed or designed out.
Kinetics may also be critical to understand and control. For PROTACs, kinetics is important to understand to get good degraders but in other cases it may be even more critical. Turning on phosphorylation or gene transcription at just the right time (and turning it off again) may be key to getting the efficacy/safety balance right.
Finally, finding novel binders to substrate and effector proteins may be needed in many cases. Options to use antibodies or nanobodies exist but delivery challenges make intracellular applications more complex so there will always be a desire to use small molecule binders. Many biophysical approaches exist for doing this (DEL is not always the best despite what many claim) but for some targets, including the classically undruggable ones, this may be a long and complex process. Any binders found must also not inhibit the intrinsic function of the effector to be useful - it can be a challenge needing to find suitable allosteric sites. We also know that, presumably due to geometrical constraints, binders to some substrate or effector sites give efficient ternary complex formation whilst others simply don’t – so having access to binders of multiple sites may also be needed.
In many ways, ligand finding remains perhaps the biggest challenge facing all forms of induced-proximity work. Many groups seem to be applying AI/ML to TPD & induced-proximity these days but, for designing cell-active molecules this is a VERY TOUGH route to go due to the complex, multi-parametric nature of the problems. However, finding ligands to hard-to-drug proteins IS a problem where the latest versions of AlphaFold and related docking and molecular optimisation algorithms could have the biggest impact.
Overall, there’s a multitude of opportunities to apply induced proximity approaches to produce therapeutics with exciting, differentiated efficacy with the potential to be commercially very successful game-changers and really help a lot of patients. PROTACs have laid out a pathway to identify and develop these agents and in some cases we can apply this directly to related proximity-based therapies. However, other applications will require careful thought and design, building on PROTAC experience. They may ultimately prove to be the most exciting but there will be work to do and great science to discover on the way.

© 2023-2024
All rights reserved. Janus Drug Discovery Consulting Ltd
We need your consent to load the translations
We use a third-party service to translate the website content that may collect data about your activity. Please review the details in the privacy policy and accept the service to view the translations.