
When acid + amine does not equal amide. Diverse TPD linkers now at your fingertips
1st January 2024
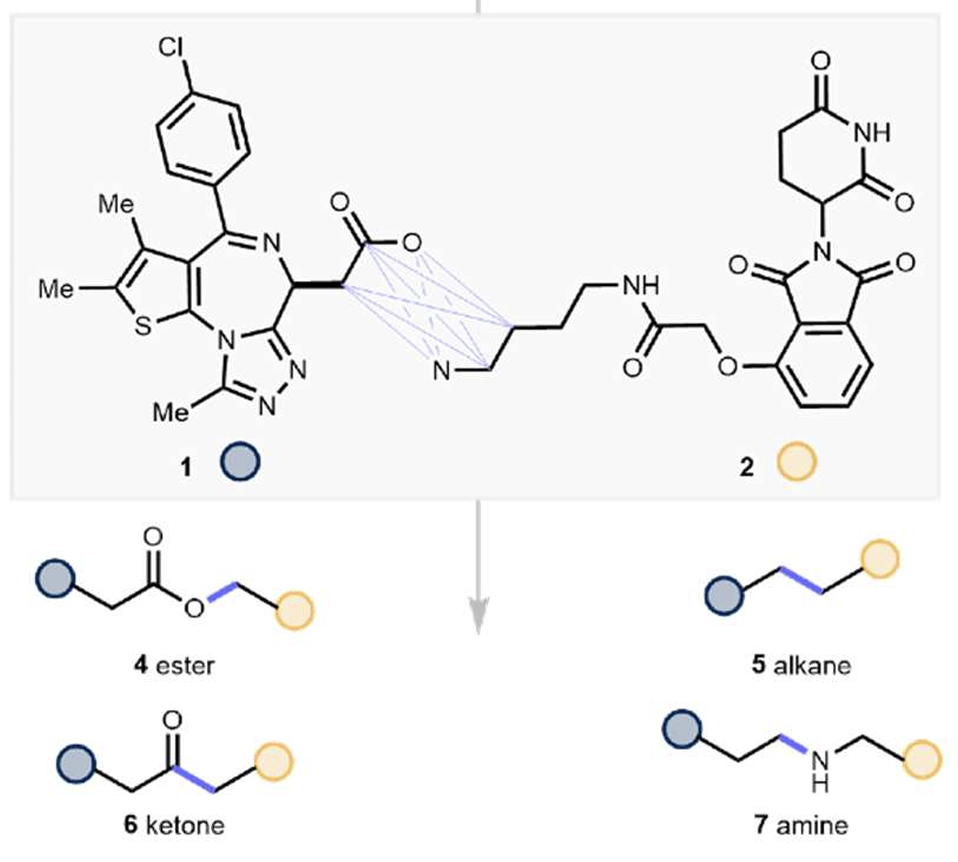
When making bifunctional molecules for TPD and other applications there can often be limited scope to vary the “end” ligands which bind the proteins to be brought into proximity. The linker however is where the magic happens and where the biological potency and drug-like properties can really be optimised so there’s a need for flexible chemistries which allow a wide range of linkers to be assessed quickly and easily. The recent U Michigan/Janssen preprint (here) does just that and is a great addition to the TPD synthetic toolbox.
Designing the linker of bifunctional molecules is often the most challenging part of the optimisation process. A good linker gives you highly potent, in vivo active molecules suitable for advancement into the clinic however, making small changes, often has profound and not always predictable effects on the properties of the molecule. A “preferred” or “ideal” linker has often been sought which can simply be introduced to any bifunctional molecule to predictably give potent, in vivo active degraders but such a linker has not yet emerged – or, if it has been discovered, it is a jealously-guarded secret.
The reality is that different bifunctional molecules need different linkers – different lengths, degree of flexibility/rigidity, polarity etc – so a wide range of molecules need to be prepared and tested experimentally to find the best ones.
Many companies are now using machine learning to try to predict optimal linker design and the results of this are eagerly awaited. Of all the medicinal chemistry problems to apply ML/AI to, PROTAC design is perhaps one of the most challenging: as good cell activity requires the simultaneous introduction of good cell uptake, binary binding, ternary complex formation, correct complex orientation, ubiquitin transfer and metabolic stability. It is a complex, multi-parametric problem where many of the key steps themselves do not have good training data and cannot easily be modelled or predicted.
Against this backdrop then, there has been much investment, with good reason, in high throughput experimentation (often using reactions directly interfaced to the biological assay) so large numbers of potential degraders can be made and tested quickly.
Beyond early PEG-based linkers (which are often metabolically sub-optimal) and click triazole-based linkers (which also often don’t seem to work well), many linkers rely on amides due mainly to the ready availability of acids, amines and mild conditions needed to combine them.
Looking at the published structures of clinical phase PROTACs however, those with most experience in the field use amides sparingly and instead look to introduce weakly basic amines, heterocycles and/or careful conformational restrictions to give linkers which confer the best properties. Amide linkages may therefore be suboptimal in many cases due to eg the presence of high polarity and a hydrogen bond donor which could limit membrane crossing potential or else susceptibility to protease-linked metabolic cleavage or other issues.
Methods which therefore can take acid and amine building blocks which are available in huge diversity form the usual chemical suppliers and combine them to make non-amides will be of high interest to the bifunctional field.
This is exactly what the Janssen team and Tim Cernak (previously of Merck so also understands the needs of medicinal chemists well!) group have achieved.
Using acid and amine precursors under a range of conditions in 10uM reaction scale, they produced:
- Esters - via pyridinium salt of amine + acid + base+ additive
- Alkanes - via pyridinium salt of amine + acyl fluoride of acid + nickel catalyst/ligand + Bronsted acid + manganese + additive
- Ketones - via pyridinium salt of amine + acyl carbonate of acid + nickel catalyst/ligand + manganese + additive
- Amines - via amine + acid + catalyst + phenylsilane
In all cases, the yield of product under preferred conditions was ~20-50%. This yield, which can undoubtedly be improved for specific cases of interest by further optimisation, is more than enough to give a few precious milligrams for biological testing – the sole deliverable at this first stage of drug discovery.
What is particularly impressive is the chemoselectivity and tolerance of the methods to the other functionality present in the JQ1-based and pomalidomide-based coupling partners which include numerous nucleophilic Lewis bases, an acidic proton and other potentially electrophilic, hydrolytically-labile functions. Many powerful synthetic methods work well on simple substrates but fail completely in the face of such a wide range of potentially reactive groups as is typically seen in bioactive molecules. The Michigan/Janssen teams should be congratulated for demonstrating success of their methods on these challenging substrates which augers well that the approach can also be applied on a broad set of substrates.
Indeed, the range of reactions also worked well to prepare VHL PROTACS showing good tolerance to alcohols and other functional groups also.
Although the manuscript is of highest interest due to the chemical methods it demonstrates, it also includes some interesting linker SAR conclusions. The authors chose to use the well-explored BRD4 system to assess degradation efficiency. While TPD-relevant observations with BRD4 are frequently not transferable to other targets, it serves a useful purpose as a probe target here.
It’s interesting to see that, in terms of cellular degradation efficiency, esters, ketones and alkyl linkages compare favourably to the archetype amide. The secondary amine on the other hand shows much weaker degradation, possibly as a result of poorer cell uptake as well as unfavourable interactions with charged BRD4 surface residues. The systematic effects of this linker variation on parameters such as in vivo metabolic stability, oral absorption, membrane efflux or protein binding would also be of great interest.
Overall, methods like these can allow a wide range of chemically diverse linkers to be prepared easily from a small set of precursors without excessive investment in synthetic chemistry time - representing a big advance for the field. Admittedly, it remains to be seen if a single set of optimal reaction conditions can be used each time or whether the system will need to be optimised for each set of substrates (using high throughput experimentation arrays) but it’s likely most substrates will be successful (eg yield >10%) under some conditions most of the time.
Perhaps we’ll now see a reduction in the number of less interesting PEG-containing or amide-containing linkers in PROTAC publications as there’s no now excuse for not also trying to introduce these kinds of non-amide functional groups in addition to the simple amines. If the current methods can also be extended to also access the kinds of linkers seen in clinical phase PROTACs, then its utility will be only increased further.

© 2023-2024
All rights reserved. Janus Drug Discovery Consulting Ltd
We need your consent to load the translations
We use a third-party service to translate the website content that may collect data about your activity. Please review the details in the privacy policy and accept the service to view the translations.